Overview
At the NAO, we use short-read Illumina technology as the gold standard for untargeted sequencing of RNA viruses in wastewater because it provides deep sequencing at a consistently low cost per base, enabling the reliable detection of rare viral genomes amid a high background of human and bacterial nucleic acids. To complement this, we incorporate Oxford Nanopore Technologies (ONT) sequencing, which offers faster sample-to-sequencing turnaround, more complete genome assemblies, and rapid verification of flagged threats. However, the same wastewater characteristics that suit Illumina—low viral abundance, degraded RNA, and high background—present challenges for ONT’s long-read, shallower sequencing. These challenges can be addressed by optimizing cDNA synthesis and PCR strategies to boost viral yield, reduce background noise, and improve both long-read sequencing performance and genome recovery.
We evaluated the performance of three ONT workflows for sequencing RNA viruses in wastewater: ONT’s Native Barcoding protocol and two versions of their Rapid metagenomic sequencing protocol: the published version of the Rapid SMART protocol (“Orig-RPDSMRT”), and a custom modification (“Cust-RPDSMRT”). Orig-RPDSMRT outperformed the other workflows across key metrics, including the highest viral fraction, lowest human background, broadest viral diversity, longest viral read length, most complete viral genome coverage, and one of the lowest duplication rates. Notably, it recovered the greatest number of unique viruses—an especially encouraging result given the primary objective of the workflow is efficient viral RNA recovery. While its total read count was the lowest, this limitation is outweighed by its superior performance in all other areas. Based on these results, we recommend Orig-RPDSMRT as the preferred ONT workflow for sequencing RNA viruses in wastewater.
A complete step-by-step protocol is available on Protocols.io.
Methods
To determine a preferred method for recovering viral RNA genomes from this complex sample type, we compared three ONT-based workflows: ONT’s Native Barcoding protocol and two versions of their Rapid metagenomic sequencing protocol: the original Rapid SMART protocol (Orig-RPDSMRT) and a custom version (Cust-RPDSMRT) (Figure 1). Native Barcoding is a PCR-free ligation-based method which we paired with oligo(dT) and randomly primed cDNA synthesis. Both Rapid SMART protocols incorporate a template-switching oligo (TSO) during reverse transcription. Orig-RPDSMRT uses only random primers, while Cust-RPDSMRT employs both oligo(dT) and random primers, better aligning with the primers previously used in Native Barcoding. Both Rapid SMART protocols include PCR barcoding. Additional details are provided in the Appendix.
We applied each of these workflows to the same set of wastewater influent samples:
- Sample A: Sampled 2025-06-03 from a sewershed in South Florida (through a collaboration with Helena Solo-Gabriele’s lab at the University of Miami)
- Sample B: Sampled 2025-06-04 from Deer Island Wastewater Treatment Plant (North System) in Massachusetts
- Sample C: Sampled 2025-06-04 from Deer Island Wastewater Treatment Plant (South System) in Massachusetts
After sequencing, we performed basecalling and demultiplexing with Dorado v0.9.1, using the [email protected] model. We used our publicly available mgs-workflow pipeline (2.9.0.4) to perform basic QC and taxonomic profiling and to identify vertebrate-infecting viral reads, then used custom Python scripts to perform additional analysis and produce plots. We then performed computational analyses of the reads from each protocol, examining (1) vertebrate-infecting viral reads; (2) taxonomic composition and human fraction; (3) read counts and lengths; and (4) duplication rate.
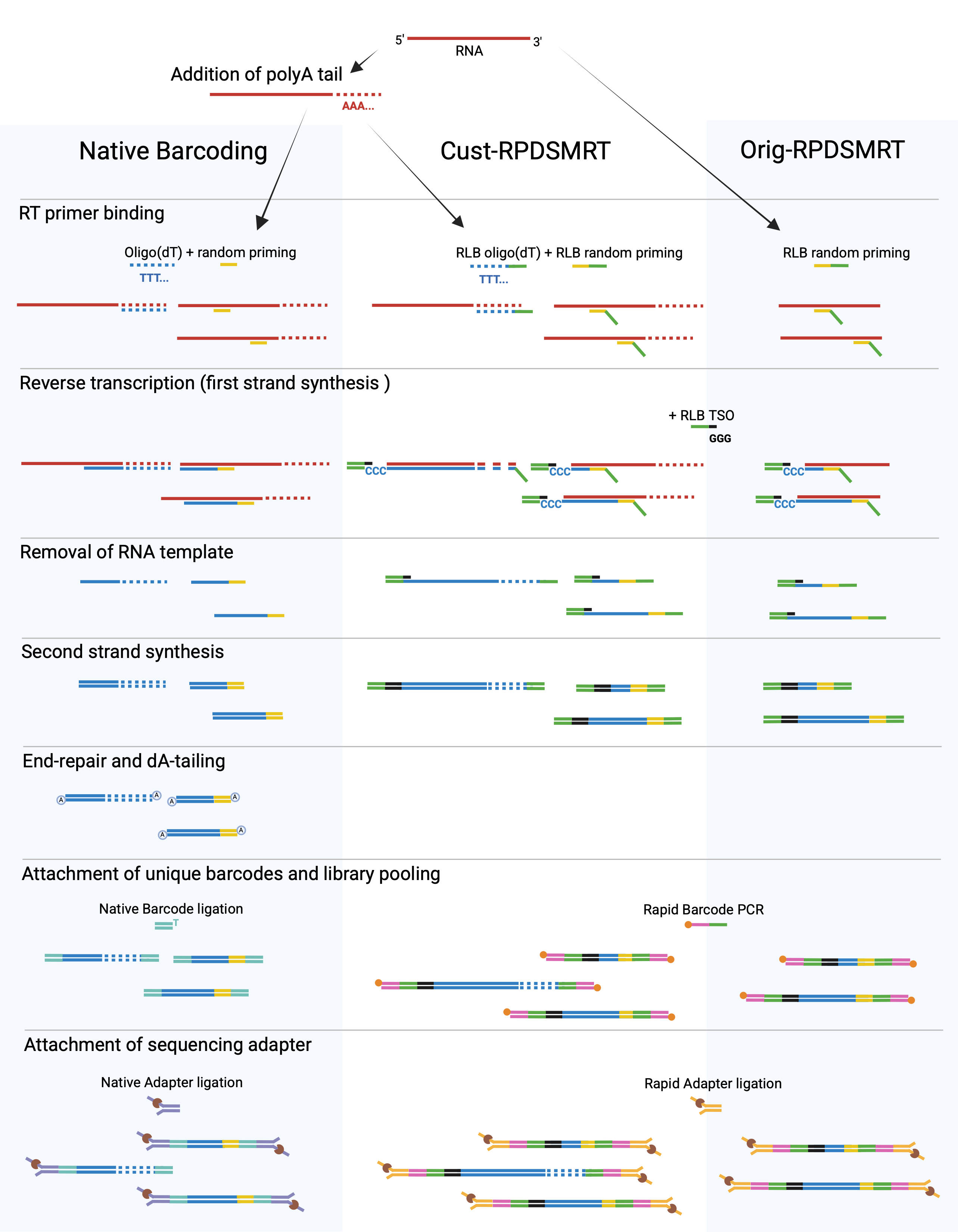
Figure 1: Stepwise comparison of the three ONT-based workflows. Created in BioRender. Smilansky, V. (2025) BioRender.com/rz511lq
Results
Overall, we find that the “Orig-RPDSMRT” protocol outperformed others on key metrics, and that 25 PCR cycles outperformed 15 PCR cycles. We are most interested in studying vertebrate-infecting viruses: Orig-RPDSMRT has the highest proportion of these taxonomies of interest, allowing us to monitor them more cost-effectively. It also has overall higher-quality data, with longer reads and lower duplication rates compared to the other protocols.
Vertebrate-infecting viral reads
We examined each protocol to check which gave the highest vertebrate-infecting virus (VV) content (Figure 2). We first looked at the VV read fraction (top panel); Orig-RPDSMRT with 25 PCR cycles had by far the highest read fraction of all the protocols. We next checked two measures of unique viral material: genome coverage (total viral base pairs, middle panel), and number of unique viruses found (bottom panel). These measures should not be affected by duplication rate. Orig-RPDSMRT with 25 PCR cycles looked better by all measures than both Native Barcoding and Cust-RPDSMRT.
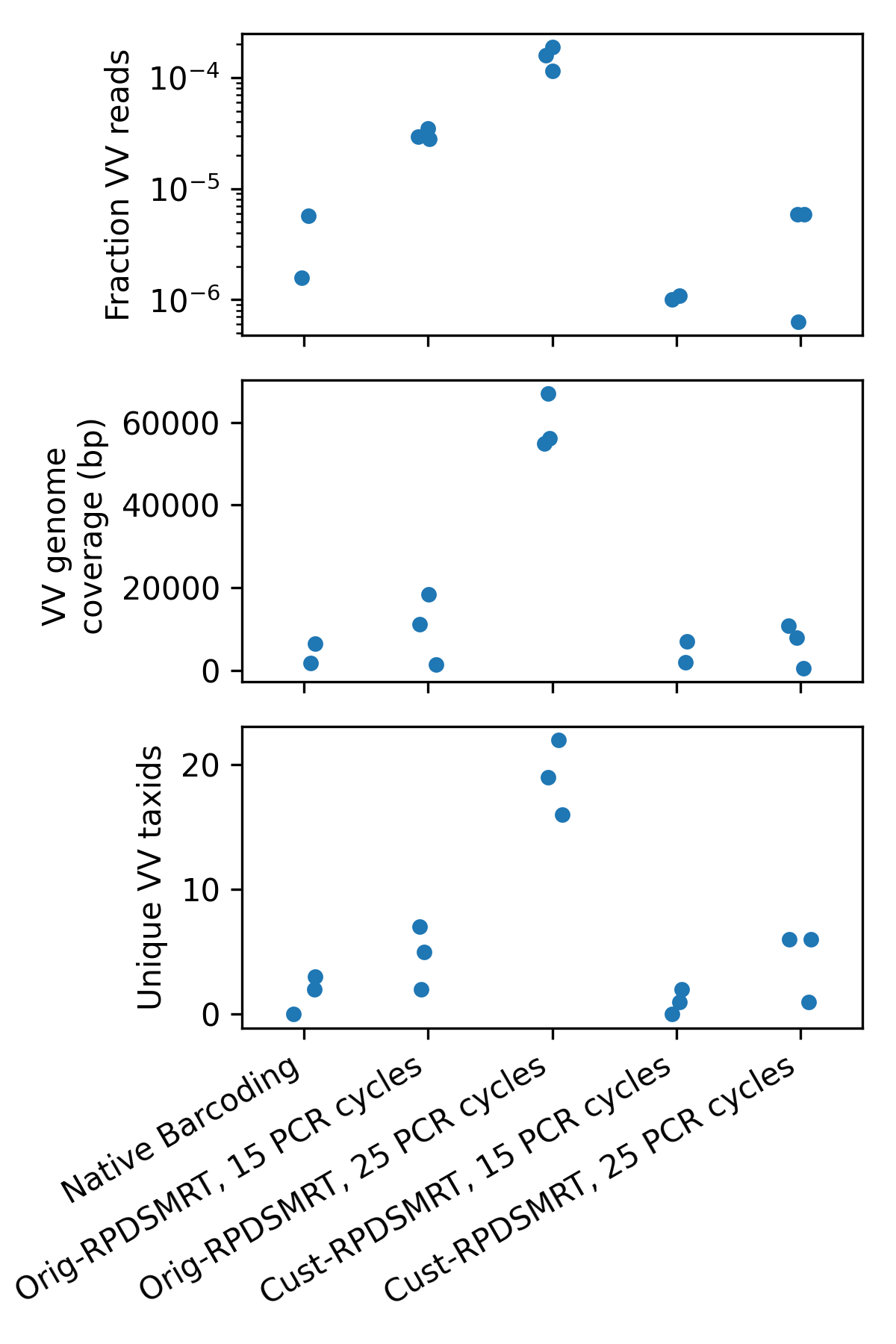
Figure 2: Vertebrate-infecting virus (“VV”) content, as measured by fraction of reads from vertebrate-infecting viruses (top panel), total coverage of VV genomes (middle panel), and unique VV taxonomic IDs found (bottom panel). Each point represents a sample/protocol combination.
In general most of the viruses found with other protocols are also found with Orig-RPDSMRT (25 PCR cycles); the only notable one missing is salivirus. We have one salivirus read in Sample A with both the “Orig-RPDSMRT, 15 PCR cycles” protocol and the “Native Barcoding” protocol; and one salivirus read in Sample B with the “Cust-RPDSMRT, 25 PCR cycles” protocol; these read counts are low/sporadic enough that this is most likely just random variation.
By contrast, “Orig-RPDSMRT, 25 PCR cycles” found many common wastewater viruses that were not found with any other protocol, including coxsackievirus, enterovirus, norovirus GII, sapovirus, and rhinovirus. Based on VV content, “Orig-RPDSMRT, 25 PCR cycles” is clearly the best protocol.
Taxonomic composition and human fraction
We next looked at the taxonomic composition of each sample (calculated by the mgs-workflow pipeline using kraken2, bracken, and custom scripts) (Figure 3). The taxonomic composition of Orig-RPDSMRT (especially with 25 PCR cycles) looks very promising! It has the largest viral fraction, suggesting that this protocol is the most successful at capturing our pathogens of interest. And it has a quite low fraction of “reads removed by cleaning” (i.e., adapter-contaminated or very short reads that are filtered out by the pipeline and cannot be taxonomically classified).
By contrast, Cust-RPDSMRT looks potentially problematic–it has the highest fraction of “reads removed by cleaning” and of eukaryotic reads. To follow up, we looked specifically at the fraction of reads mapping to the human genome (Figure 4); we want this to be as low as possible. As we can see, this is much higher with the Cust-RPDSMRT protocol than with either of the other protocols.
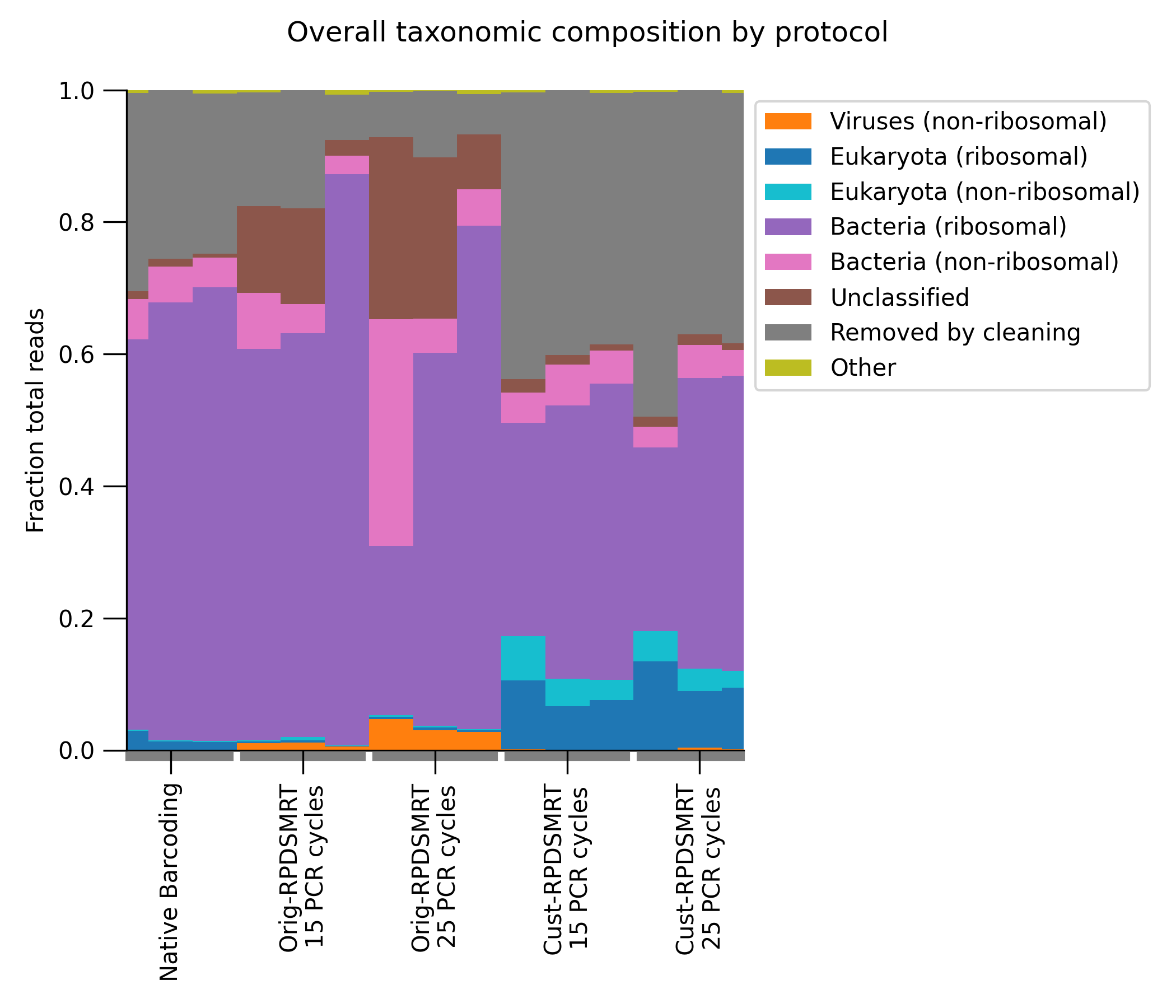
Figure 3: Taxonomic composition of samples with each protocol
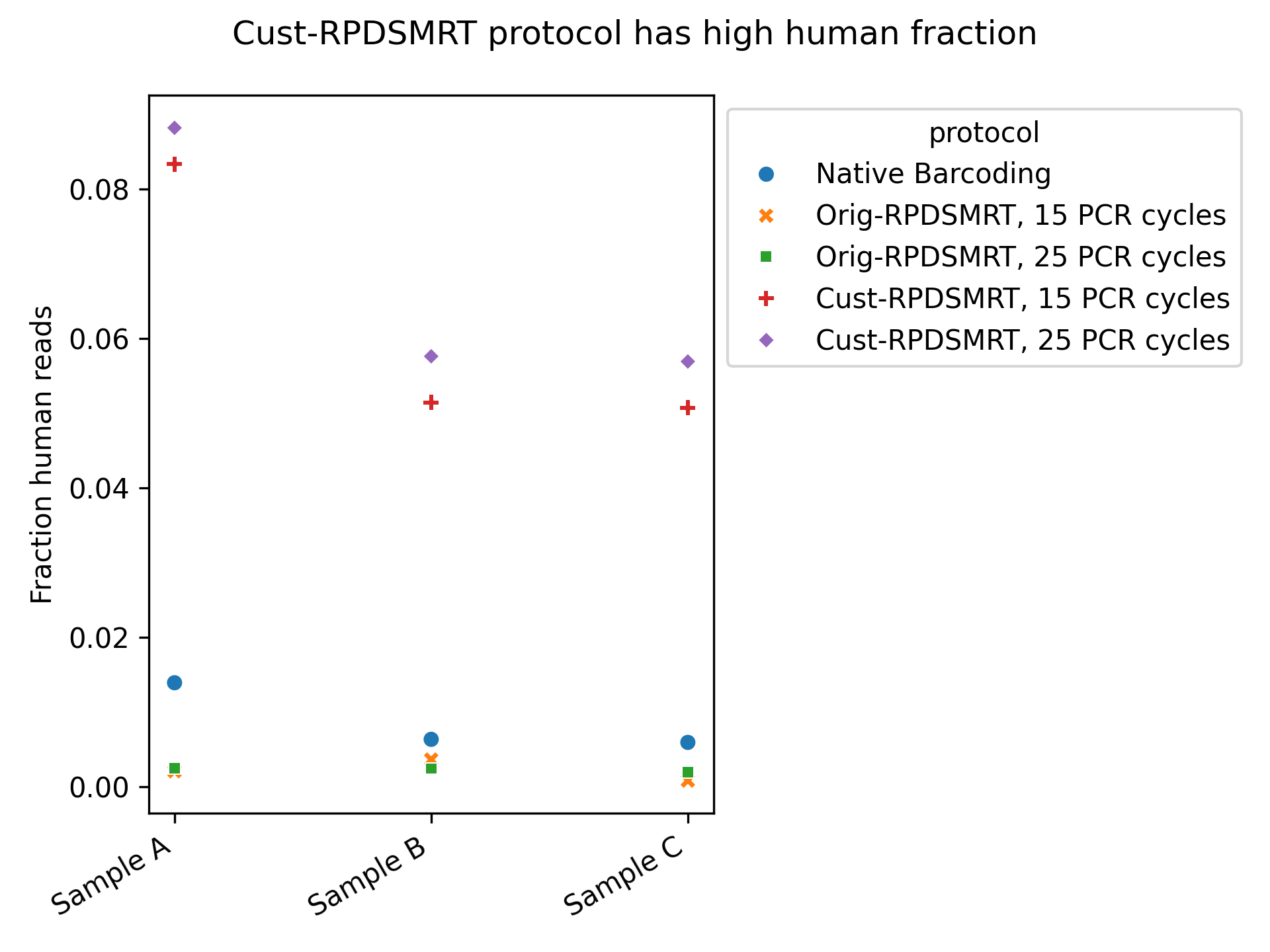
Figure 4: Human read fraction for each sample/protocol combination
Read counts and lengths
We next checked read counts (Figure 5) and read lengths (Figure 6) for each protocol. We focused on comparing read counts within the two different variations of the Rapid SMART protocol.1 We see that Orig-RPDSMRT has the lowest read counts but the longest reads, while Cust-RPDSMRT has the opposite (high read counts but short read lengths).2 As we would expect, more PCR cycles boosts read count.
The read counts for Orig-RPDSMRT with 15 PCR cycles are concerningly low, but read counts and lengths look acceptable for all other protocols.
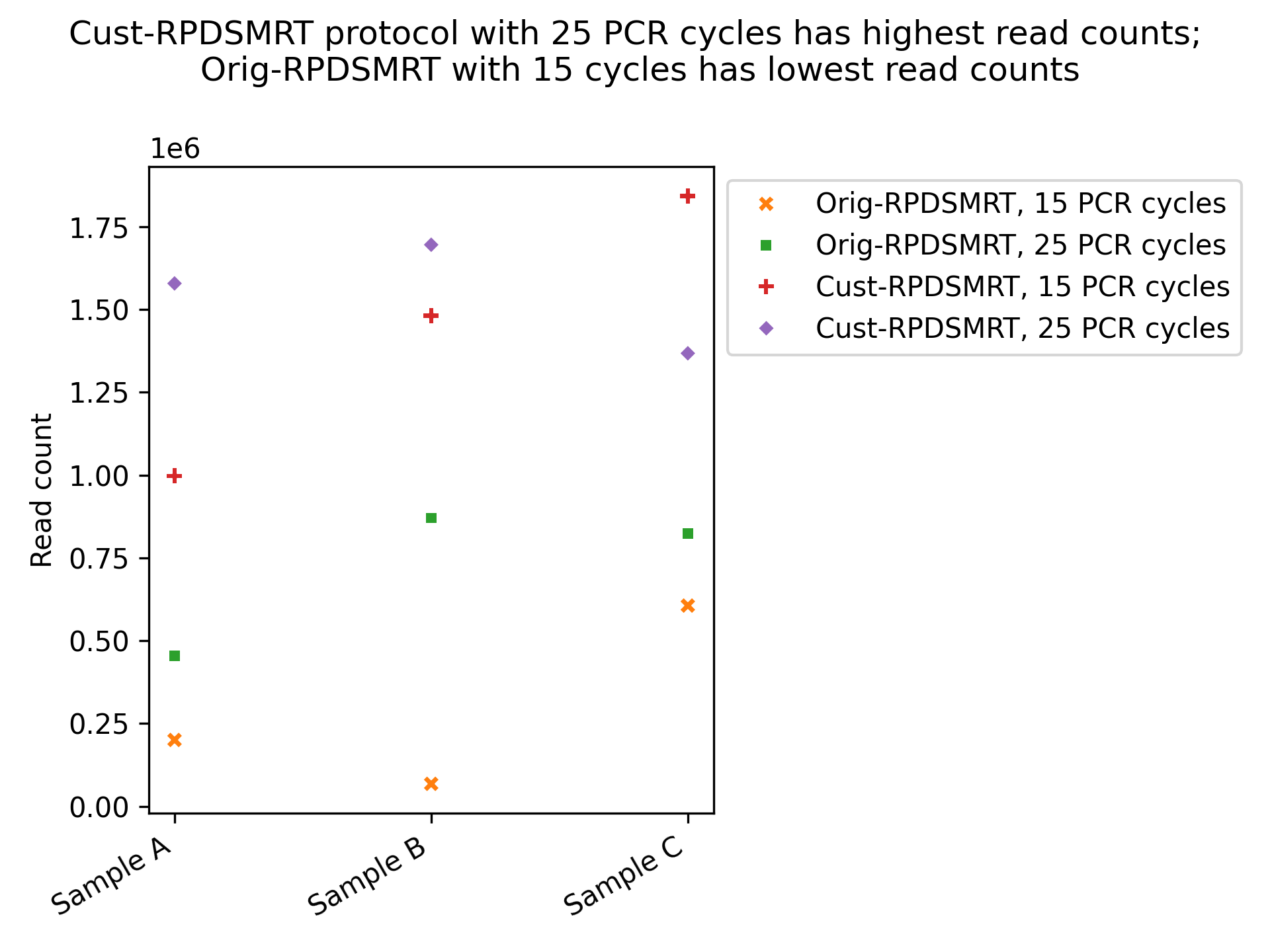
Figure 5: Read counts for each sample/protocol combination
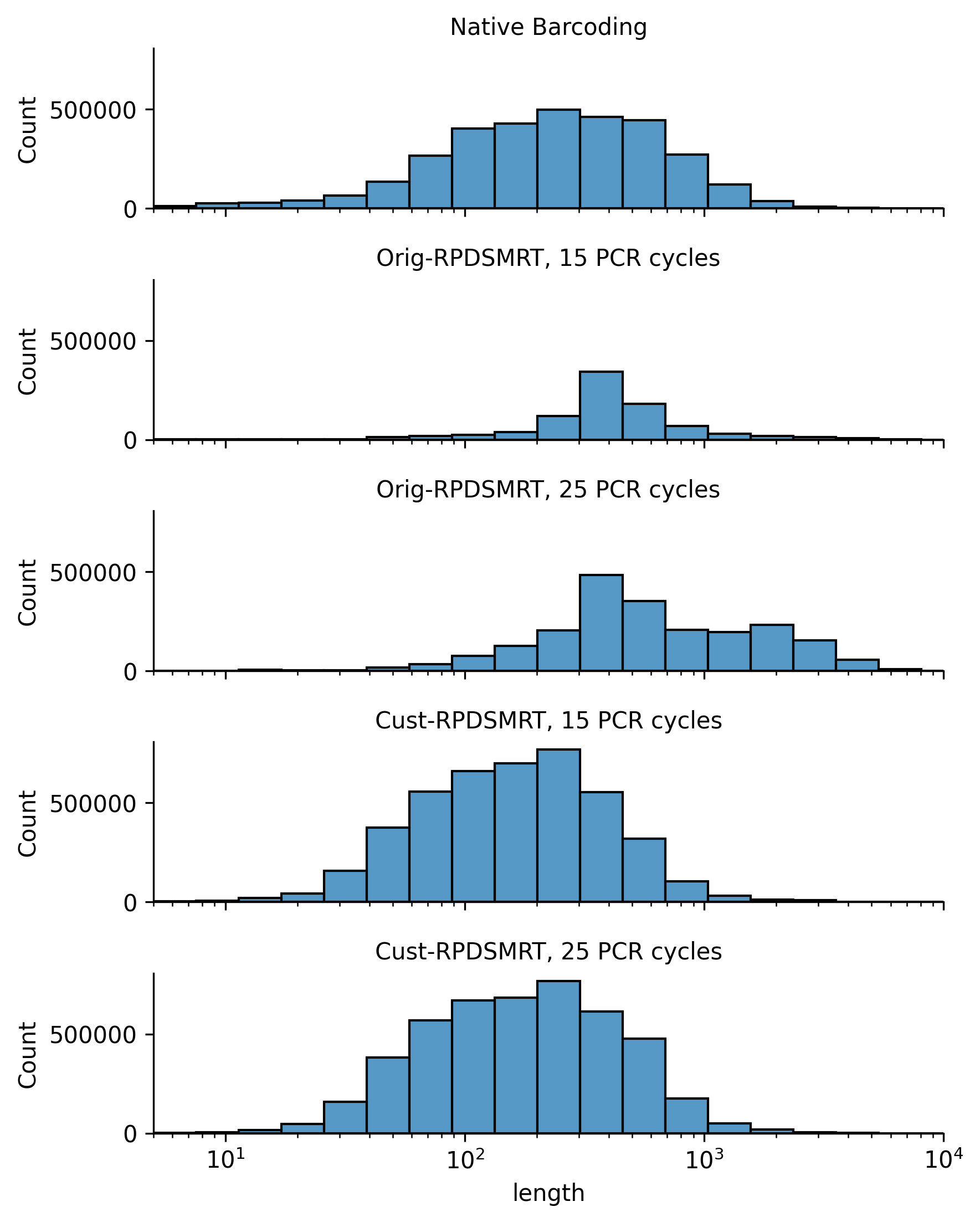
Figure 6: Read length histogram for each protocol
Duplication rate
Finally, we look at duplication rates, as measured by FASTQC (Figure 7). We downsampled each sample to ~65,000 reads, the lowest number of reads observed in any sample, to avoid confounding by differing read counts. We also tried excluding Orig-RPDSMRT with 15 PCR cycles (which had anomalously low read counts, Fig. 1) and downsampling to ~500,000 reads. We can see that Orig-RPDSMRT has the lowest duplication rate; 15 and 25 PCR cycles give similar duplication rates.
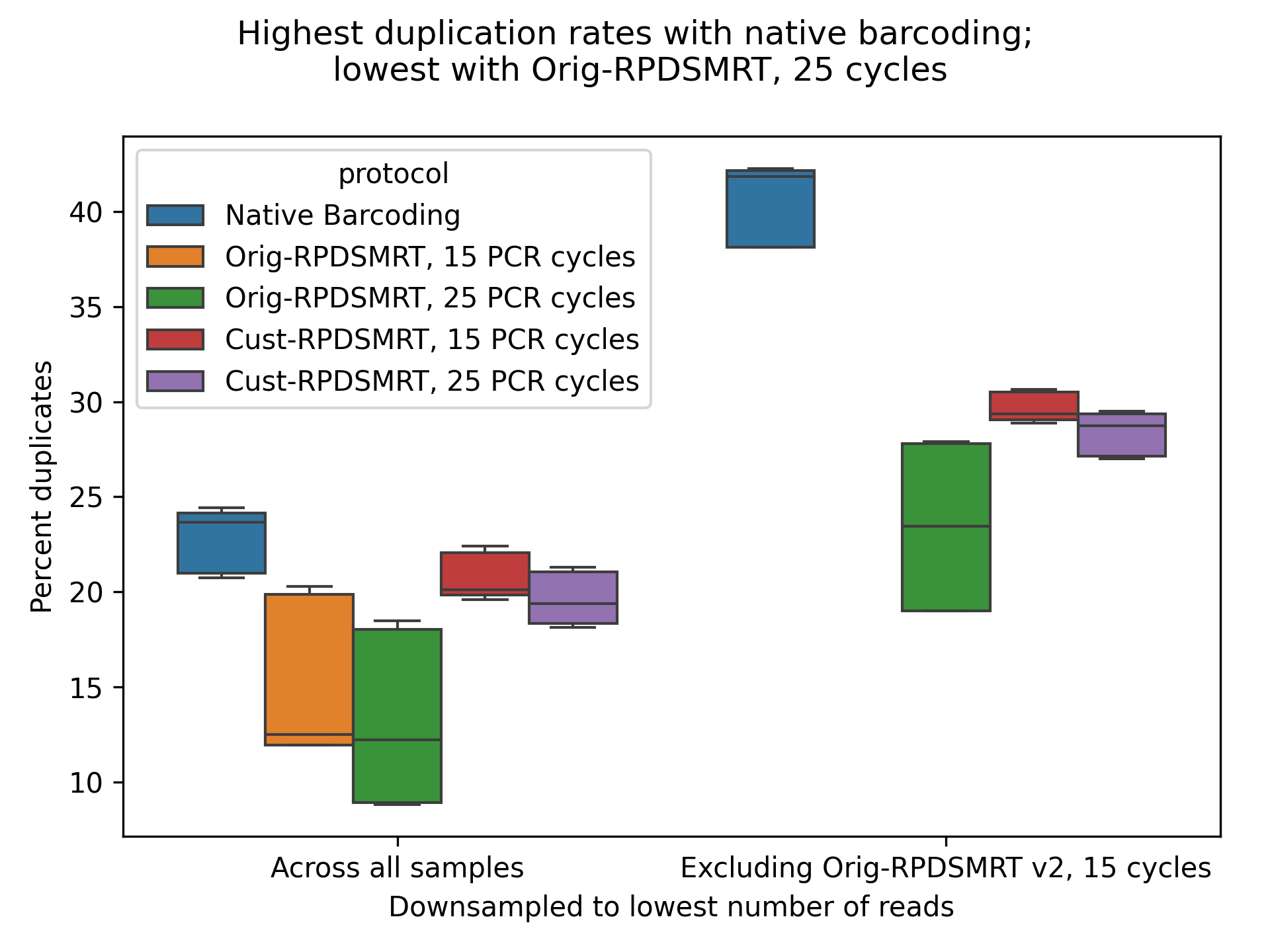
Figure 7: Duplication rate for each protocol, as measured by FASTQC on downsampled subsets of the data.
Conclusion
We evaluated ONT’s Native Barcoding protocol and two variants of their Rapid metagenomic protocol—Cust-RPDSMRT (random + oligo(dT) priming) and Orig-RPDSMRT (random priming only)—for sequencing RNA viruses in wastewater. Each Rapid SMART method was tested with 15 and 25 PCR cycles. Orig-RPDSMRT with 25 cycles delivered the best overall performance across key metrics: it yielded the highest viral fraction, lowest human background, longest viral reads, broadest viral diversity, most complete genome coverage, and lowest duplication rate—despite producing fewer total reads. These advantages, particularly in viral diversity and genome completeness, make Orig-RPDSMRT our recommended workflow. Our recommendation is further substantiated by recent large-scale, non-public wastewater experiments that sequentially applied the Native Barcoding and Orig-RPDSMRT workflows, confirming that Orig-RPDSMRT reliably produces high-quality datasets. Routine implementation of Orig-RPDSMRT is expected to enhance our biosurveillance capacity by enabling rapid and comprehensive validation of flagged viral threats. Its fast turnaround and operational simplicity enable scalable deployment in field-based public health monitoring, facilitating outbreak tracking and timely decision-making.
Appendix
Raw wastewater was preprocessed and extracted according to the NAO protocol Concentration and nucleic acid extraction of viruses from wastewater influent V.3. Extracts were treated with DNase to reduce background. DNased extracts were divided into three aliquots: A1 samples were processed with the Native Barcoding workflow, A2 with Cust-RPDSMRT, and A3 with Orig-RPDSMRT.
Historically, we have favored Native Barcoding to avoid the additional fragmentation caused by ONT’s Rapid chemistry, which is poorly suited for wastewater samples containing degraded nucleic acids, as further fragmentation can produce fragments too short for effective sequencing. Rapid kits generally use a transposase to fragment and tag dsDNA with adapters, which are either pre-barcoded or barcoded via PCR. In contrast, ONT’s Rapid metagenomic sequencing protocol avoids the transposase step by adding adapters during cDNA synthesis; these adapters are complementary to the fixed sequence on Rapid Library Barcodes (RLBs) and are incorporated via modified reverse transcription primers (Table S1).
To align with our standard Native Barcoding workflow, we adapted this approach by adding a similarly modified oligo(dT) primer and included a polyadenylation step prior to cDNA synthesis (Cust-RPDSMRT). We also tested ONT’s recommended version, which omits both polyadenylation and oligo(dT) priming (Orig-RPDSMRT). Both protocols use an RLB template-switching oligo (TSO), which binds to Cs added at the 5′ end of the cDNA by reverse transcriptase, enabling the enzyme to extend and incorporate the RLB adapter. This captures full-length cDNA and streamlines downstream amplification. For both Rapid workflows, we used the NEB LunaScript® RT Master Mix Kit (Primer-free).
For the Native Barcoding protocol, we first polyadenylated the DNased extracts, then synthesized cDNA using oligo(dT) and random primers provided in the NEB LunaScript® RT SuperMix Kit to maximize viral genome recovery and cDNA fragment length. The resulting cDNA underwent end-repair and dA-tailing (i.e., adding a single A overhang to cDNA ends) to enable ligation of T-overhangs on Native Barcodes. This was followed by library normalization, pooling, and final ligation of the Native sequencing adapter. We loaded 207.6 ng of pooled libraries onto a previously used PromethION flow cell with 2,179 available pores. The pool was sequenced for 43 hours and 46 minutes, generating 2.96 Gb of data.
Both A2 and A3 aliquots underwent Rapid Barcode PCR using 25 and 15 cycles—fewer than the recommended 30—to account for the higher input from omitting ONT’s host depletion step, and to test whether reducing cycles could lower duplication rates. The libraries were then normalized and pooled, and ligated with Rapid sequencing adapters. We loaded 118.8 ng of pooled libraries onto a previously used PromethION flow cell with 2,384 available pores. Sequencing ran for 43 hours and 13 minutes, yielding 9.63 Gb of data.
The cDNA synthesis approach that we used for Native Barcoding yielded a relatively high amount of cDNA that was sufficient for ONT’s recommended input of ~130 ng of sample per barcode ligation (Table S2). Both Orig-RPDSMRT and Cust-RPDSMRT yielded sufficient cDNA for the recommended 5 ng input for the Rapid Barcode PCR (Table S2). Cust-RPDSMRT yielded higher concentrations, which could be due to the combination of random + oligo(dT) priming.
As expected, the 25-cycle PCR produced more cDNA than the 15-cycle PCR. Before pooling, all samples were normalized to match the lowest concentration. Several of the 15-cycle reactions yielded very little product, resulting in a total input of 180 ng for bead cleanup—below ONT’s recommended 800 ng. Nevertheless, the final concentration of 10.8 ng/µL was adequate for loading, particularly given the use of a previously run flow cell with reduced pore availability.
Supplemental tables
Table S1: Primers used for first strand cDNA synthesis in the Rapid SMART workflows
| Primer name | Sequence |
|---|---|
| RLB RT 9N | 5’-TTTTTCGTGCGCCGCTTCAACNNNNNNNNN-3’ |
| RLB RT 20T-VN | 5’-TTTTTCGTGCGCCGCTTCAACTTTTTTTTTTTTTTTTTTTTVN-3’ |
| RLB TSOmg | 5’-GCTAATCATTGCTTTTTCGTGCGCCGCTTCAACATmGmGmG-3’ |
Table S2: Comparison of three ONT-based workflows using wet lab metrics
| Sample ID | Protocol | cDNA conc. (ng/uL) | ng for PCR | PCR cycles | PCR conc. (ng/uL) | ng for ligation | Pool conc. (ng/uL) |
|---|---|---|---|---|---|---|---|
| Sample A | Native Barcoding | 42.5 | NA | NA | NA | 136 | |
| Sample B | Native Barcoding | 21.8 | NA | NA | NA | 136 | |
| Sample C | Native Barcoding | 18.3 | NA | NA | NA | 136 | |
| 17.3 | |||||||
| Sample A | Orig-RPDSMRT | 2.7 | 5 | 15 | 0.51 | 15 | |
| Sample B | Orig-RPDSMRT | 34.1 | 5 | 15 | 0.309 | 15 | |
| Sample C | Orig-RPDSMRT | 2.79 | 5 | 15 | 1.33 | 15 | |
| Sample A | Orig-RPDSMRT | 2.7 | 5 | 25 | 11.7 | 15 | |
| Sample B | Orig-RPDSMRT | 34.1 | 5 | 25 | 8.36 | 15 | |
| Sample C | Orig-RPDSMRT | 2.79 | 5 | 25 | 11.5 | 15 | |
| Sample A | Cust-RPDSMRT | >60 | 5 | 15 | 1.56 | 15 | |
| Sample B | Cust-RPDSMRT | >60 | 5 | 15 | 1.35 | 15 | |
| Sample C | Cust-RPDSMRT | >60 | 5 | 15 | 2.36 | 15 | |
| Sample A | Cust-RPDSMRT | >60 | 5 | 25 | 27.2 | 15 | |
| Sample B | Cust-RPDSMRT | >60 | 5 | 25 | 9.37 | 15 | |
| Sample C | Cust-RPDSMRT | >60 | 5 | 25 | 11.2 | 15 | |
| 10.8 |
Footnotes
Pooling fewer libraries increases sequencing output per library, while larger pools reduce per-library yield due to increased competition for available pores. Because Native Barcoding was performed with fewer libraries per pool, direct comparisons with the Rapid SMART workflows are challenging. Variability in flow cell performance further complicates this comparison, as some flow cells yield more data regardless of pool composition. For these reasons, Native Barcoding was excluded from our read count comparisons.↩︎
The tradeoff between read count and length, may reflect our normalization approach–we normalize libraries by DNA concentrations, which doesn’t account for the different average molecule sizes between protocols. This normalization effect may have reduced the read counts from the Rapid SMART–original.↩︎