The pipeline described in this post is a team effort with significant contributions from Harmon Bhasin and Ryan Teo. Thanks to Mike McLaren and Jeff Kaufman for reviewing drafts of this post.
Summary
When the NAO’s detection system flags a suspicious sequence, genomic context is key in determining whether this is a real threat. The sequencing reads containing the sequence are a good start, but in the tricky cases we need to assemble additional reads beyond these initial observations into a larger contig — contiguous genomic sequence — containing the suspicious region. While assembly is a widely studied problem, standard metagenomic assemblers don’t scale well to the volume of data we’re now processing. We’ve implemented a computational pipeline that efficiently searches through large volumes of sequencing data to iteratively extend contigs outward from initial seeds. This outward assembly pipeline can generate target organism contigs from tens of billions of read pairs in under an hour.
Motivation
The NAO operates a wastewater monitoring system designed to detect engineered stealth pandemics. Every week we generate tens of billions of sequencing reads from municipal wastewater samples and analyze them to look for signs of genetically engineered human-infecting viruses. For example, our chimera detection pipeline flags sequencing reads that contain a junction between “human infecting virus” and “something else,” which is a sign of potential genetic engineering. Our sequencing data currently consist of Illumina paired-end 150-base-pair reads; the reads in a pair typically overlap and jointly only cover around 200 base pairs total. The ~200bp merged reads flagged by chimera detection are often too short to determine which organism(s) the sequences come from and what their biological function might be — in short, how concerned we should be. In these cases, our next step is to assemble at least a couple hundred base pairs of genomic context on both sides of the junction.
Conventionally, genomes are recovered from untargeted metagenomic sequencing data via full metagenomic assembly — an entire sample is jointly assembled into contigs, or multiple samples are coassembled together. Unfortunately, this standard approach is inefficient for our early pathogen detection work. Individual samples rarely contain sufficient reads to build meaningful context around a flagged sequence. Instead, we must aggregate reads from multiple related samples: the original sample containing the flag, other samples from the same location, temporally similar samples, samples from the same flow cell, etc. This aggregate set of potentially relevant samples — we don’t know a priori which samples contain useful reads — may contain dozens of samples with several billion read pairs each for a combined size of hundreds of billions of read pairs. Coassembly at this scale is possible only with significant investments of time, money, and computational resources, e.g. distributing the assembly over nodes of a supercomputer. Additionally, it’s not guaranteed that a large coassembly will yield contigs containing the flagged sequences, and tuning assembly parameters to increase sensitivity to rare sequences can drastically increase computational requirements.
Fortunately, we don’t need to assemble the genomes of all the organisms in wastewater, just those related to our flagged sequences. Most of the reads are irrelevant: the overwhelming majority of reads in our data correspond to unrelated organisms (e.g. non-pathogenic bacteria found in sewers). In fact, because we try to detect suspicious sequences before they’re common, our flags correspond to organisms with very low relative abundance in the wastewater data — typically much lower than one in a million reads. This means that the actual assembly we care about — assembling the genomes that contain the flagged regions — requires very little data, perhaps hundreds or low thousands of read pairs. If we could find the relevant reads, assembly would be fast.
Outward Assembly
The NAO’s outward assembly pipeline iteratively builds contigs outward from a starting seed1 such as the 50 base pairs surrounding a flagged chimeric junction. The basic intuition behind the algorithm is simple:
Start with the seed as your initial contig. Then iteratively:
- Find all read pairs that share a kmer (short sequences of exactly k consecutive bases) with your contigs.
- Assemble these read pairs into contigs.
- Filter the resulting contigs to those that contain the seed.
Repeat the iterative process until either maximum iterations are reached or the contigs do not grow from one iteration to the next.

Ideally, each iteration’s search step pulls in additional reads that enable assembling longer contigs than in the previous iteration — and these contigs in turn contain kmers that can be used to pull in new reads in the next iteration. Eventually, an iteration will fail to identify reads not already identified by prior iterations, and the assembled contigs will match the prior iteration; at this point the algorithm terminates.

Assembling a spike-in
A previous blog post described our experiments, in collaboration with Marc Johnson’s lab at the University of Missouri, with engineered viral particles spiked into samples of municipal wastewater. The spike-ins are derivatives of a laboratory HIV strain with an inserted region, and they provide natural test cases for outward assembly both because we know the underlying genome we’re hoping to assemble and because the spike-in genomes contain junctions flagged by our chimera detection pipeline.
As a test, we revisited this sequencing data and outward assembled the v530 spike-in genome starting from the first junction between the HIV genome and the inserted DsRed (a red fluorescent protein) gene. Our seed was the 40-mer symmetrically spanning the junction: the last 20 base pairs of HIV genome before the DsRed gene concatenated to the first 20 base pairs of the DsRed gene.
The sample containing the spike-in was sequenced to a depth of 668 million read pairs, of which only 329 were from the spike-in genome. Thus the relative abundance of spike-in in the data is 4.9e-7: comparable to pathogen relative abundance in wastewater, and a good candidate for outward assembly.

In this simple test case, outward assembly identifies only a single seed-containing (or seed-overlapping) contig per iteration. For the first five iterations, the contig grows rapidly — roughly 240 additional base pairs per iteration. After the fifth iteration, however, the left edge of the contig has reached a genome region of zero coverage: none of the 329 spike-in read pairs cover this region, and outward assembly cannot assemble through such a region. Thus after the fifth iteration the contigs continue growing only to the right. The iteration 9 contig shown in Figure 3 is 1,972 base pairs long and aligns to a matching 1,972 base pairs of the spike-in reference genome with 99.5% identity. Although we didn’t assemble the entire spike-in genome, our contig covers the entire inserted DsRed gene as well as hundreds of base pairs of viral backbone on both sides of the insert — enough to deduce which gene was inserted into which virus. Generating this contig took approximately 22 CPU-hours; for comparison, assembling the full 668 million read-pair sample with MEGAHIT took 551 CPU-hours and did not produce a contig containing the seed.
Outward assembly implementation details
The NAO’s outward assembly pipeline implements the core iterative logic described above with several specializations for our use case:
Performance
The overwhelming majority of pipeline compute is spent searching for reads that contain known kmers (step A of the iterative process). We implement efficient search by streaming carefully formatted and compressed sequencing data from AWS S3. We also support arbitrarily wide parallelization of this search using Nextflow and AWS Batch.
Coverage and sensitivity
We try to detect growing pathogens as early as possible, meaning the relevant genomes have very low read coverage in our data. Thus we confront an unavoidable tradeoff:
- Observed genetic sequences attested to by very few reads are generally unreliable: they may contain sequencing errors,2 and we don’t want to assemble errors into our contigs. Ideally, we only assemble genetic sequences attested to by multiple non-duplicate reads.
- Because the (unknown) target genomes have low coverage, extending flagged sequences into long contigs may require assembling genomic regions that are covered by very few (sometimes just one) reads.
In most assembly contexts, this tradeoff is managed by setting a coverage threshold r: only assemble regions that are covered by at least r reads; r = 3 is common. Since random sequencing errors are typically observed only once or twice each, this prevents most assembly of errors into contigs. But in our case, such a high threshold is not safe — it might prevent correct assembly of a rarely observed error-free sequence. Instead, outward assembly runs multiple assemblies per iteration, varying the coverage threshold (and other parameters) between assemblies. When an assembly with a high coverage threshold (i.e. more conservative) extends the contigs from the previous iteration, we use that assembly — but when it doesn’t, we can fall back to assemblies with lower coverage thresholds.
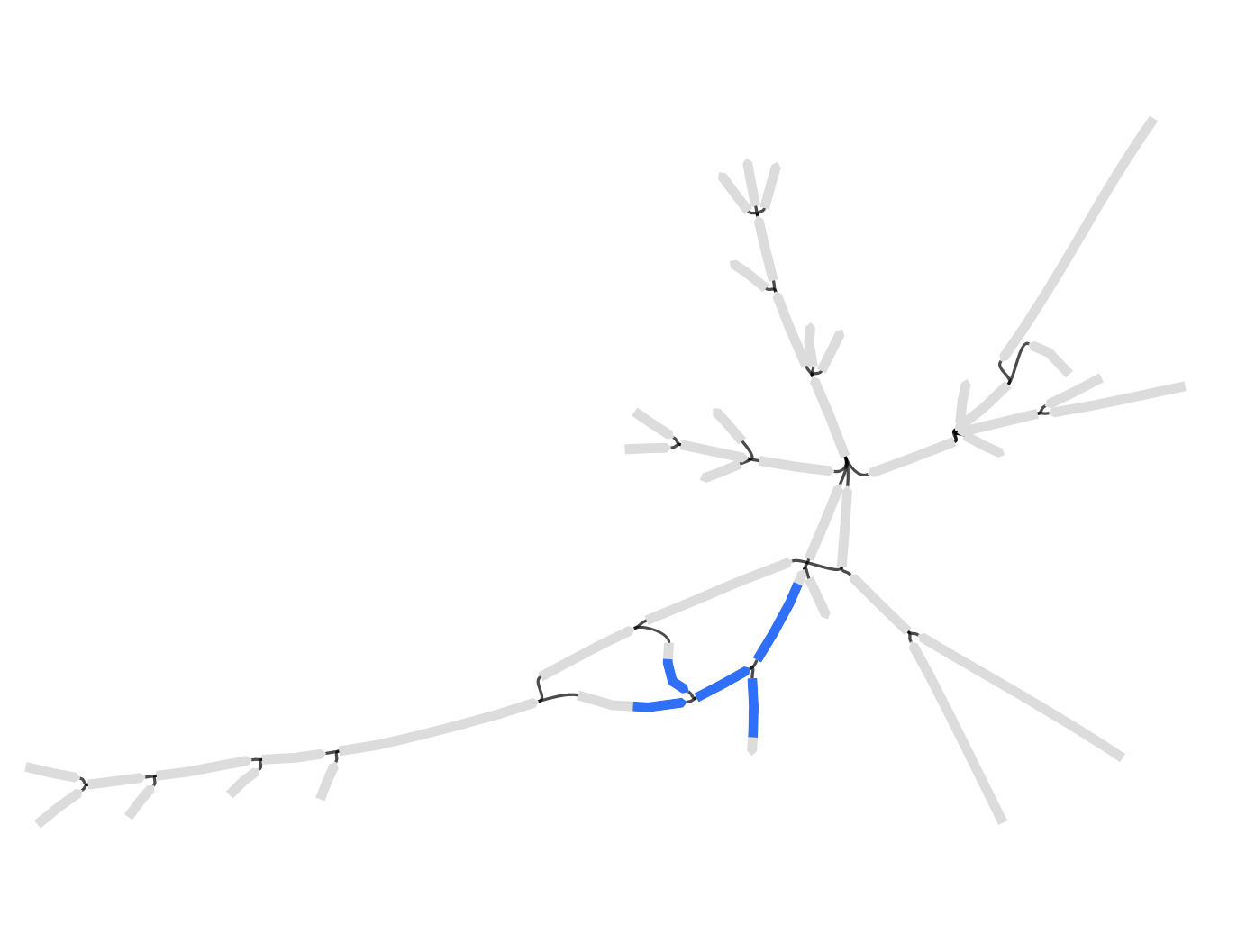
Branching and related contigs
In standard assembly, ambiguities lead to branches in assembly graphs and thus to shorter contigs. For example, if reads attest to genomes AB and AC (where A, B, and C are sequences of some length), assemblers will return separate contigs A, B, and C, since it’s ambiguous whether A is followed by B or C. But if our seed is contained in contig A, we should also retain contigs B and C, even if they don’t directly contain the seed: both are possible extensions of the seed-containing contig and may carry valuable information. The outward assembly pipeline supports identifying relevant contigs via a two-step process. First, outward assembly computes all overlaps between contigs — where the end of one contig matches the beginning of another. Then outward assembly retains all contigs that either contain the seed or overlap with a contig containing the seed.3
Configurable resources
The iterative nature of outward assembly is especially handy for our use case. Often a single iteration of outward assembly — just a few minutes of elapsed time — suffices to stitch together 150-base-pair mates into ~500-base-pair contigs. If we need longer contigs, we can let outward assembly run for more iterations or on larger amounts of input data.
Looking forward
Outward assembly is already helping us understand junctions flagged by our chimera detection pipeline. We’re also building outward assembly into an experimental reference-free detection pipeline, where outward assembly is key for classifying kmers with increasing abundance. The NAO’s outward assembly pipeline is now open source, and if you’re interested in collaborating or using outward assembly, we’d love to hear from you.
Footnotes
The ideas of assembly starting from a known seed and iterative contig extension appear in the literature, though these approaches are significantly less common than full assembly. See e.g. this review of related assembly tools.↩︎
Or otherwise don’t represent real genomes; e.g. the reads could represent technical chimeras.↩︎
More generally, any contig that either contains the seed or is connected to the seed by a chain of contig overlaps.↩︎