We have a lot to share since our April update! As always, if you have questions or see opportunities to collaborate, please let us know; we’re eager to work with others thinking along similar lines.
Wastewater Sequencing
We’ve continued expanding our collaboration with Marc Johnson’s lab at the University of Missouri to sequence wastewater influent from six additional sewersheds across four states, including Boise ID. We’ve also continued ramping up our in-house process, sequencing at Broad Clinical Labs. In Q2 the two labs have sequenced 487B read pairs (242B at MU, 245B at BCL), 101% more than in Q1. Marc has worked with Dave O’Connor at the University of Wisconsin-Madison to put together a public dashboard showing human-infecting and animal-infecting viruses in the MU-sequenced samples.
We’ve uploaded an initial batch of this sequencing data, which is available on SRA under accession PRJNA1247874, covering 170 billion read pairs from Boston MA, Chicago IL, and Riverside CA. While we have still not automated our upload process, we expect to do another round of manual uploads in Q3, which should cover approximately 250 billion read pairs.
We’ve continued our ANTI-DOTE contract work for PHC Global, sequencing weekly samples from US military facilities. While we think Illumina is generally more appropriate for untargeted wastewater sequencing than Oxford Nanopore (ONT), due to the low relative abundances of many pathogens and Illumina’s much lower cost per basepair (~$3/Gbp vs ~$300/Gbp in our testing), we’ve been performing some ONT sequencing for this contract. In Q2 we switched from ONT’s Native Barcoding protocol to a modified version of ONT’s Rapid metagenomic sequencing protocol, after experiments showing longer reads and more reads per run.
This quarter we’re going to be running some experiments combining hybridization capture with ONT sequencing to get good coverage of human-infecting viruses in municipal wastewater influent. We’re starting with Twist’s Comprehensive Viral Research Panel, which has 1M+ probes across 3k+ genomes. If we’re able to get this working well, something in this area might be a good fit for detection of novel pathogens derived from known viruses.
During our Fall 2023 collaboration with CDC’s TGS and Ginkgo Biosecurity we banked several hundred pooled airplane lavatory waste and municipal treatment plant influent extractions. We had a full set of RNA and DNA libraries prepared at MIT’s BioMicroCenter, and sequenced a representative subset. Unfortunately it appears that the high-throughput library prep process we chose did not produce RNA libraries suitable for sequencing at the depth we had hoped. We may still sequence the full set of RNA libraries at a lower depth, and are considering whether to sequence the DNA libraries (which don’t appear to have the same issue). We still have aliquots of all the original extractions, and are continuing to bank them in case trade-offs here change.
We ran several experiments attempting to replicate the protocol of Yang et al. (2020), which had reported one of the highest fractions of human-infecting viruses we’d seen in the literature. When combining their extraction protocol with our standard approach to concentration (using the Innovaprep CP) and ribodepletion (riboPOOLs pan-bacterial) the extraction protocol seems to be in the same ballpark as our protocol, but more experiments are needed.
Pooled Individual Sequencing
We’ve been scaling up our Boston-based swab sampling program. We’ve hired our first two dedicated field samplers, who go out sampling each Tuesday. This has allowed us to start collecting 100-200 swabs in a day of sampling. We’re continuing to optimize collection, and are hoping to get up to a consistent 200-300 swabs per day.
We’ve received permission to begin sampling at MBTA stations, and have started experimenting there in addition to our sidewalk testing. We are looking at expanding to additional weekdays in Q3, and if this work continues to go well we plan to ramp more, sampling each weekday during winter 2025-2026 to capture the historical increase in respiratory infections.
We published a blog post, Pooled Lab Discards for Pathogen Detection, investigating pooled lab discards as a sample type. We think this is promising, especially as a way to expand coverage to blood samples, but don’t currently have the capacity to spin up a third sample type.
Analysis of Sequencing Data
A significant effort during Q2 was updating our main analysis pipeline to better handle rare and ambiguous sequences. This has included the addition of lowest common ancestor (LCA) analysis, BLAST-validation, and improved deduplication. Together, these improvements should give us better estimates of the relative abundances of viral clades. In Q3, we will use these estimates to resume work on flagging clades whose abundances are growing.
We also extended our main pipeline to process Oxford Nanopore (ONT) data, unifying our metagenomic sequencing processing. Because ONT data differs along several key dimensions, especially longer reads at lower accuracy, the appropriate bioinformatics tools will often be different for the same goal.
Our data import system is now automated, instead of requiring people here to notice emails from our wet lab or sequencing partners and manually kick off jobs. This will help us scale up our sequencing with less labor from the dry lab team, and also reduce the time from sequencing data to result.
When short read data is imported, we now convert it to a format optimized for efficient highly parallel processing, and we’ve open-sourced the conversion tool. This data conversion is the first step of a larger effort to accelerate our detection pipelines so they can run end to end on new data in under an hour.
We tested our reference-free growth detection pipeline (described in our last updates post) on a single-city time series containing 84B read pairs, and we successfully identified growing sequences and organisms. However, in assembling these sequences we ran into scaling issues due to the number of short sequences exhibiting exponential growth. Applying reference-free growth detection to municipal wastewater at scale will require further work to increase the number of sequences we are able to dismiss prior to assembly. While we remain excited about reference-free growth detection, in Q3 we’re shifting our development focus to other detection approaches that we think we can get production-ready more quickly.
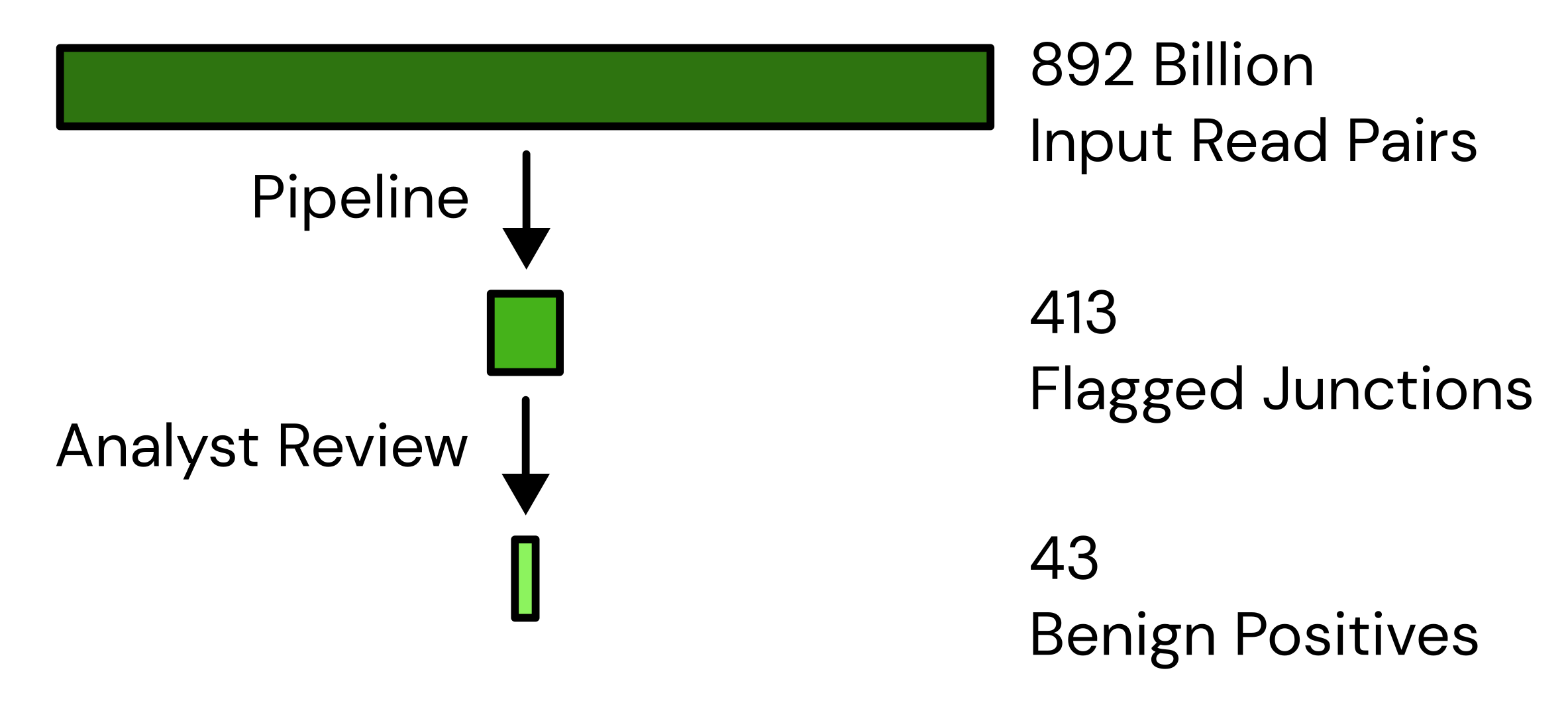
Our chimera-based genetic engineering detection system has now processed a total of 892B read pairs, of which it has flagged 413 collections of overlapping reads for human review. Most of these 413 are a range of false positives that our system is not yet sufficiently sophisticated to exclude automatically. Of these, however, 43 are what we call “benign positives”: reads from genomes that the system is correct to flag, even though they don’t represent a hazard to humans. Most of these 43 are lab contamination, primarily lentiviral and adenoviral vectors, used in other work at the Johnson Lab. We’ve also improved the flagging algorithms, increasing sensitivity while also increasing specificity, and also improved the tooling that supports the manual analysis, surfacing more relevant information to the analyst to speed up their judgements. In Q3 we plan to extend this system to automatically assemble these reads outwards, further increasing the information available to analysts.
We were able to use our swab sampling data collected in Q1 to make prevalence estimates for a range of respiratory viruses, which we could then use to get better estimates of the sensitivity of wastewater sequencing. This included substantial modeling work using the fraction of pools in which we detected a given pathogen and the number of unique reads from the pathogen to estimate population prevalence. This turned out to be quite subtle, and we are proud of our model here. We found that common cold viruses are harder to detect in wastewater than SARS-CoV-2. We are planning to continue applying this model as we ramp up nasal swab collection over Q3. We’ve written this up as a blog post, Estimating the Sensitivity of Wastewater Metagenomic Sequencing Using Nasal Swabs.
Our manuscript, Inferring the sensitivity of wastewater metagenomic sequencing for early detection of viruses: a statistical modelling study, has been accepted for publication in The Lancet Microbe, and should be out later this summer.
Organizational Updates
We’ve recently transitioned to a unified NAO leadership structure under Jeff Kaufman. This streamlined approach will allow us to focus on building, evaluating, and improving our biosurveillance system. Mike McLaren has transitioned to a senior individual contributor role where he’ll focus his deep expertise on solving our most critical detection challenges.
We’ve hired Jake Lloyd as a part-time field sampler and lab assistant for our pooled individual sampling work. Jake is a biotechnology student at Northeastern University, and previously interned at Pioneering Medicines and Solarea Bio.
Evan Fields was invited to speak at EA Global London, where he presented on the NAO’s work and how it fits into the broader biosecurity landscape. Jeff Kaufman spoke on a panel at the Berlin Biosecurity Dialogue, as part of CSR’s Global Biosecurity Accelerator, discussing how metagenomics can help fill diagnostic and surveillance gaps. On July 31st we’ll be co-hosting another Accelerating Biosecurity Networking Event with Ginkgo Biosecurity.